(Formerly #76)

 One of the cardinal points of interaction and interface between copper <> iron is that the proper absorption of iron in the duodenum of our gut requires heme iron and/or reduced ferric iron (found in non-meat food sources) to enter the enterocyte via a key protein pathway called DMT-1, as known as Divalent Metal Transporter-1. That’s the front door into the 1st tissue that receives the iron in our gut, aka the enterocytes. But it’s the back door where the real action seems to take place, especially as it relates to iron.
One of the cardinal points of interaction and interface between copper <> iron is that the proper absorption of iron in the duodenum of our gut requires heme iron and/or reduced ferric iron (found in non-meat food sources) to enter the enterocyte via a key protein pathway called DMT-1, as known as Divalent Metal Transporter-1. That’s the front door into the 1st tissue that receives the iron in our gut, aka the enterocytes. But it’s the back door where the real action seems to take place, especially as it relates to iron.At that other end, the enterocytes back up to the bloodstream, and for iron to get out of the enterocytes, and into the bloodstream, there are three critical factors to contend with:
- Another doorway, called Ferroportin, aka FPN.
- A doorman, called Hephaestin, (HEPH) which is a copper-dependent, ferroxidase enzyme that transforms ferrous (Fe2+) iron into ferric (Fe3+) iron so it can bind to transferrin; and
- A regulator, called Hepcidin, (HPN) which is called the Iron Regulatory Hormone, but it’s also called the “Inflammation Hormone!” as Hepcidin’s production is triggered by the presence of either iron and/or a set of inflammatory cytokines, IL-1, IL-6 or TNF-a.
And when hepcidin shows up consistently, this peptide causes the degradation of that back doorway and the iron egress gets stopped, thus leading to the build-up of iron in the gut. Please know this process of inflammation-driven iron retention is what leads to many gut issues, like Leaky Gut, SIBO, Crohn’s, Colitis, etc.
These mechanisms to “catch & release” iron are the classical understanding of how iron “gets into the gut,” and either it “gets out” via the presence of copper-driven hephaestin, or it “gets stuck” via the increased prevalence of iron, and thus hepcidin, in the 1st line of tissues in our digestive system.
And all is good with this ^^^^ description above, but it overlooks a glaring issue: So if copper is needed to enable iron to get out of the gut, then how in the world does iron build up in tissue, all over the body if we’re all lacking bioavailable copper?
It’s a really important question and one of my Copernican students in Group #5 asked me this very question just the other day.
My initial reaction to this sincere question was one of frustration, as expressed by my initial response: “We don’t need to know that, just accept that the process of iron build-up happens…” Hardly a proper nor a complete response, and thankfully, her question just kept grinding away at me. I couldn’t explain with certainty how this iron dysregulation and subsequent accumulation was actually happening outside the gut, given that the iron was obviously not stuck there, even though copper is likely in a deficit position.
Then, by the grace of God, I found the following breakthrough study:
Matak, P. et al. (2013). “Copper Deficiency Leads to Anemia, Duodenal Hypoxia, Upregulation of HIF-2a and Altered Expression of Iron Absorption Genes in Mice”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3610650/pdf/pone.0059538.pdf
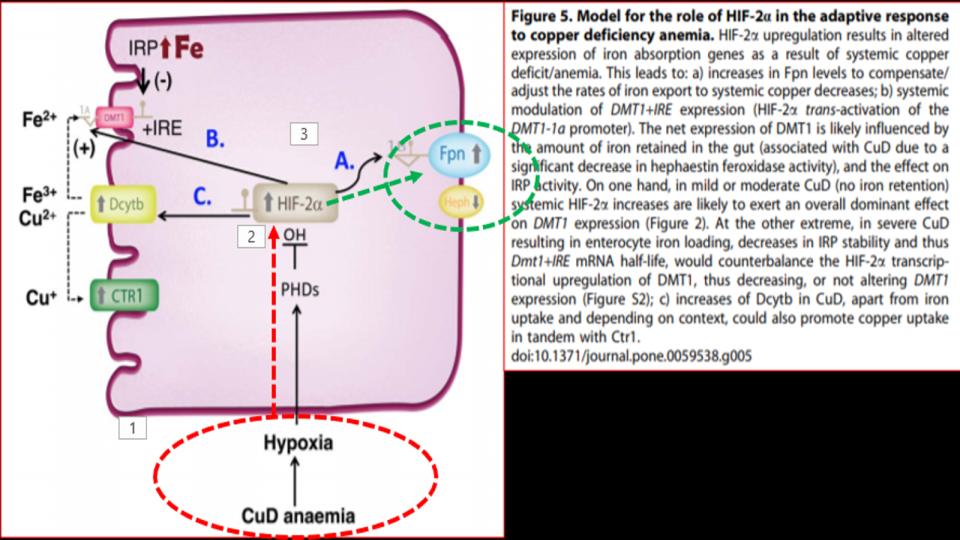
Presented at the 1st Figure (Labeled “Fig 5” at the top of this post) is the key dynamic. What we see there as you follow the diagram from the bottom up and over to FPN (on the right side) is that in the presence of a copper-deficient organism, in this case rodents, is that copper deficiency leads to hypoxia – a profound discovery, which then affects the function of a key protein called HIF-2a, as known as Hypoxia Inducible Factor-2alpha, which is a powerful protein that responds to the “lack of oxygen” signal, and triggers the opening of that key FPN doorway to allow iron out of the enterocyte and into the bloodstream.
Before we go any further, a quick digression. “Hypoxia,” as it turns out, is a double-entendre:
- “Low” oxygen implies low levels of iron, given that iron is key to “carrying” oxygen in the blood. Essentially, 70% of the body’s iron (hemoglobin) is nothing more than an “oxygen waiter!”
However,
- When you read Matak P et al (2013) you discover that low copper (or lack of bioavailable copper) also causes “Hypoxia,” as the activation of O2 in Complex IV (which is copper-dependent) is vital to transforming that O2 into 2H2O to release 3 Mg-ADPs that go over to Complex V to make the primary energy molecule, Mg-ATP. Thus, we are now learning that “hypoxia,” (which implies “low” oxygen) can also mean “low activation of oxygen” due to a lack of bioavailable copper. That is a major distinction in how that term is used and understood, clinically.
So then, what’s the significance of this study? We learn that “copper deficiency” triggers duodenal hypoxia which increases the expression of HIF-2a, which then causes:
- Increased expression of Ferroportin
- Increased expression of DMT1, thereby allowing more iron into the enterocytes
- Increased expression of DcytB, Ferric reductase, also allowing more iron
While this article is silent on this issue, the action of erythropoiesis, of making new blood cells is also copper-dependent. The significance of all of this is that we’re seeing first hand that the process of making new blood and managing the movement of Iron has great dependence on bioavailable copper, contrary to conventional thinking or popular websites.
And then lightning struck as I started to study the key footnotes to this section of the study!
[17] Evans et al, 1973, (No abstract!) “Effects of iron on copper metabolism and copper on iron metabolism in rats.” Am J Physiol 224: 514–518.
[18] Reeves et al, 2004, “Copper Deficiency Reduces Iron Absorption & Biological 1/2-life in Male Rats” J Nutr 134: 1953–1957
[Bonus] Reeves et al, 2005, “Copper Deficiency Leads to Lower Iron & Hephaestin”
[19] Williams DM, Cartwright GE et al, 1976, “Role of Copper in Mitochondrial Iron Metabolism” Blood 48: 77–85.
Please know, this third article may be among the most important articles that I’ve ever read about the critical dynamics that take place between copper <> iron in the synthesis of new blood.
What is particularly important with this study is that it relates to the dynamics that are occurring inside the mitochondria of the blood-building-erythroid cells, which reside in the bone marrow. And what’s noteworthy, is that these key cells have the greatest avidity for iron that enters our body of all the cells in our bodies.
What you’ll see below at “Table 5” is a striking dichotomy between “normal” and “copper-deficient” mitochondrial function. There are striking differences in levels and levels of activity, as you’ll note.
Contrary to popular opinion, outside of MAG FB Group or the Root Cause Protocol website, copper is a major and understood player in the actions of the mitochondria, especially the mitochondria of these critical erythroid cells that are creating new blood 24/7.
Copper is essential for three key steps in the process to make these new blood cells: 1) make energy; 2) make {Fe-S] Clusters, and 3] make Heme.
And as Table 5 highlights, it is clear that a lack of bioavailable copper has a profound negative effect on the mitochondria’s ability to perform these recognized metabolic tasks.

Furthermore, as Figure 2 (below) goes on to amplify these points, you’ll note exactly where the breakdowns are occurring in this erythroid-driven process. And one of the most important and often little discussed steps is the conversion of ferric (Fe3+) iron into ferrous (2+) iron so that this latter form of iron can be naturally dropped into the center of the heme protein by the ferrochelatase enzyme, as known as heme synthesis. Note that this key enzyme only works in the presence of bioavailable copper, and that heme is best when iron is in the ferrous form.

What is notable in this study by Williams, Cartwright et al (1976) are their sterling conclusions to drive home the central importance of copper to properly regulate iron’s role in this bone-building process:
“Four defects in iron metabolism have now been delineated in copper-deficient swine:
(1) impaired synthesis of heme from Fe(III) and protoporphyrin;
(2) impaired mobilization of iron from reticuloendothelial cells to transferrin;
(3) impaired mobilization of iron from hepatic parenchymal cells to transferrin; and
(4) impaired absorption of iron from the gastrointestinal tract. [1]
We propose that the first of these defects are due to a deficiency of cytochrome oxidase. The second and third defects are due to a deficiency of ceruloplasmin. [3] The nature of the defect in the mucosal cell has not yet been defined, but it could be due to a deficiency of both cytochrome oxidase and ceruloplasmin.
Thus, it is apparent that at least two copper proteins, cytochrome oxidase, and ceruloplasmin, are involved in the movement of iron.”
This set of findings, albeit with swine, are a compelling set of critical conclusions that demonstrate not only the interplay between copper <> iron, but the obvious conclusion that copper appears to have a more regulatory role, and iron a more passive role, in this critically guided process, known in clinical circles as erythropoiesis.
So, why is this all so important?
1) We cannot make new blood without the active and regulatory role of copper
2) We need optimal levels of copper to prevent the activation of HIF-2a, and the unexpected release of iron into the bloodstream, and ultimately into the tissues all over the body.
And finally,
3) We need optimal levels of copper to prevent the build-up of iron in the tissues that then leads to the relentless tug-of-war that invariably happens inside our bodies between our metabolism and the pathogens living, replicating and thriving on that excess, unbound iron in our tissues and organs. Again, Pasteur forgot to tell us that the pathogens thrive on that iron buffet!
This last point is driven home by the compelling study noted below:
Skaar, Eric P, (2010). “The Battle for Iron between Bacterial Pathogens and Their Vertebrate Hosts”
The accompanying graphic (Fig 1) presented below demonstrates the impact of having excess iron in the systemic mix… Given that all pathogens – Bacteria, Fungus, Virus, and Parasites – require iron for their existence, it’s noteworthy that so many people are being labeled “anemic”.
It appears from this ongoing set of studies in this post, there is a glaring demonstration of the essential role of copper and it’s nutrient partner, real vitamin-A, retinol. Again, this fat-soluble vitamin is instrumental in making our copper bioavailable, and thus enabling our iron to be properly usable and functional and thus avoid the inevitable production of “accidents with oxygen,” that invariably happen inside a low copper body.

- A LOT LESS declarations of “Anemia!”
- A LOT LESS Supplementations with Iron and Iron infusions
AND
- A LOT LESS potential for pathogenic build-up to foster oxidative stress, inflammation (whether by virtue of iron and/or pathogen-generated lipopolysaccharides) and then a cascade of symptoms and labels.
Folks, this Cu<>Fe dynamic is the head of the stream that leads to the relentless confusion about the “status of iron” in the blood vs. the tissue, and the oft-recommended need for supplemental iron that compounds the metabolic dysfunction creating a myriad of symptoms.
Please know that there are notable differences between iron levels found in the liver/spleen tissue and the iron levels found in the bone marrow. And therein lies the problem.
The levels of iron found in the blood are not representative uniformly of the other two. And subsequent posts on Iron Toxicity will drill into how this distinction is so important and why this is the origin of the mineral dysregulation that affects our metabolism.
We covered a lot of important ground today. Hopefully, you followed the switchbacks. And I would advise any and all that are dealing with the “anemia” label to re-read this post and please print out a copy for their practitioner to read, as well
“A” votre sante!
Morley M. Robbins
https://www.facebook.com/groups/MagnesiumAdvocacy/posts/1871997332868327/